
The singular goal of this article is to flesh out the role of gluten in your diet by walking you through the scope of the issue, the biological data and plausible mechanisms, and then help you fully think through the pragmatic aspects of gluten-free diets.
Part 1: About Gluten
The Scope of the Issue
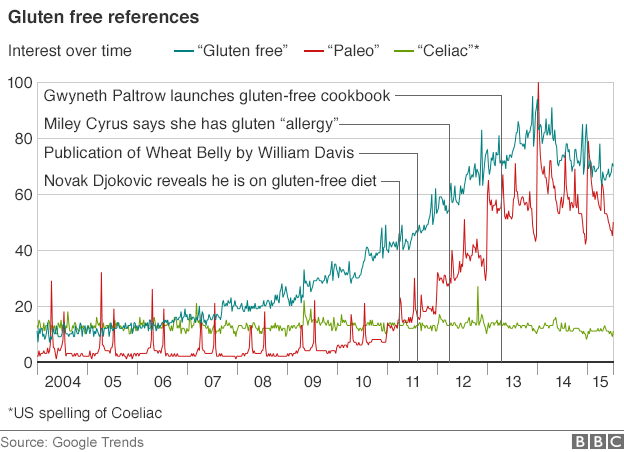
There has been a substantial increase in the interest of gluten and the application of gluten free diets. As you will see below, much of this interest can be attributed to the popularization of diets like the Paleo Diet and popular press books like Wheat Belly than by some meteoric rise in celiac disease (CD).
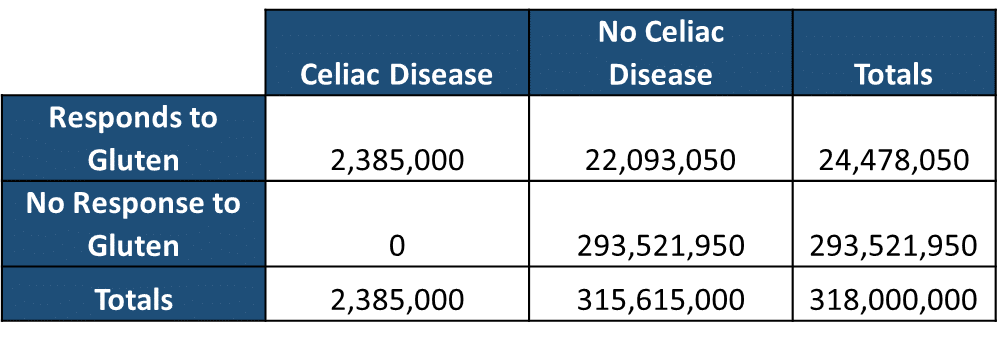
As disease prevalence goes CD is fairly common; it effects between 0.5%-1.0% of the entire human population, with about 1 in 133 Americans suffering from the disease. This accounts for roughly 2-3 million people in the United States. In addition to those suffering directly from CD, there is another class of people who do not have CD react to the ingestion of gluten, this is known as non-celiac gluten sensitivity (NCGS). From most reports in the literature, between 5-9% of people without CD report adverse reactions to gluten*; the symptoms that people with NSGS report to have occur across such a wide spectrum (foggy mind”, depression, ADHD-like behavior, abdominal pain, bloating, diarrhea, constipation, headaches, bone or joint pain, and chronic fatigue). When take the entire American population, estimated at 318 million people, and we break down the numbers of people with and without CD, and those with and without adverse reactions to the gluten protein, it is clearly easy to see the actual numbers of people who may truly require a gluten free diet (Table 1).
When you hold these numbers juxtapose to the explosion of the interest in gluten and gluten free diets since 2009 (See Figure 1) it is clear that this rise in interest is better explained by the popularization of diets like the Paleo Diet and popular press books like Wheat Belly than by some meteoric rise in celiac disease (CD). There is clearly a dichotomy between the interest and use of gluten-free diets and the medical necessity of them; this is most likely due to the lack of understanding of exactly what gluten is, how it is involved in weight gain and disease, and a combination of people cashing in on a fad and people honestly not really knowing what they are talking about. The topic is quite complex and requires sorting through some things in detail. At the end of this article we should be able to answer the following questions: how big of a deal is gluten and should you be on a gluten free diet?
What is Celiac Disease and Non-Celiac Gluten Sensitivity
Celiac disease (CD) is a life-long gluten-sensitive autoimmune disease of the small intestine and CD diagnosis is based on presence of predisposing genetic factor human leukocyte antigen (HLA) DQ2/8, with positive biopsy and serological antibodies upon gluten contained diet (1). CD commonly appears in early childhood, with severe symptoms including chronic diarrhea, abdominal distension, and failure to thrive. In many patients, symptoms may not develop until later in life, when the disease symptoms include fatigue, diarrhea, and weight loss due to malabsorption, anemia, and neurological symptoms (2). Interestingly, CD does not typically manifest at birth and is usually “triggered” by something. The development of CD requires the genetic predisposition (specific HLA haplotype) and an exogenous trigger (sometimes gluten) or even viral infections or other intestinal tissue damage. NCGS presents with similar symptoms to gluten exposure without the presence of the predisposing genetic factor human leukocyte antigen (HLA) DQ2/8 and no serological antibodies upon exposure to a gluten containing diet. A gluten allergy (or wheat allergy) presents differently than both CD and NCGS and is an immune reaction in which the body develops IgE antibodies against gluten or wheat proteins.
What is Gluten?
Gluten is often thought of as a singular protein; however, it is actually a mixture of proteins found wheat and other evolutionarily related grains. It is a composite of prolamins like giadin, glutenin, hordeins, secalins, and avenins. Gluten, as traditionally thought of is a combination of gliadin and glutenin protein. As it is currently understood gliadin appears to the protein most causal in the disease process associated with CD and NCGS.
Part 2: Hard Science
Proposed Mechanisms on non-celiac gluten sensitivity.
CD is relatively straight forward. The body produces an immune response to the ingestion of gluten that increases gut permeability and causes villious atrophy and flattening of the cells that line your intestines. Removal of gluten from the diet usually ameliorates most symptoms.
NCGS is a much more complicated, nuanced issue as there appears to be some merit to the condition. One of the leading and most compelling hypothesis surrounding NCGS is increased intestinal permeability through a gliadin-CXCR3-Zonulin mechanism. Essentially, gliadin, a gluten protein, interacts with the CXCR3 receptor in intestinal cells, releasing a protein called zonulin which increases intestinal permeability. This intestinal permeability is what causes NCGS symptoms.
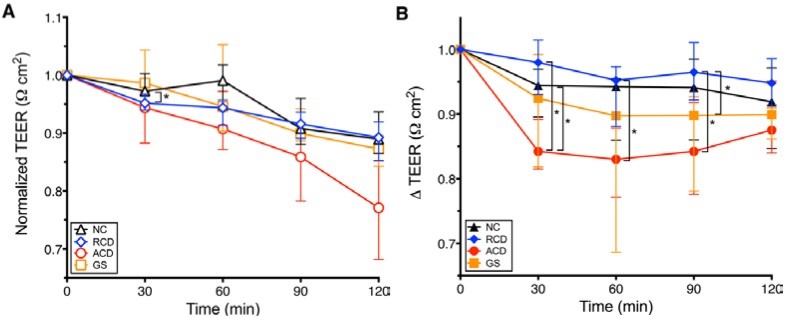
There are in vitro and animal studies that work out this mechanism in detail (3, 4). In ex-vivo human duodenal biopsies from people with active celiac disease, people with remitted celiac, non-celiac gluten sensitivity, and non-celiac controls, increased intestinal permeability after gliadin exposure occurs in all individuals. Following gliadin exposure, both patients with gluten sensitivity and those with active celiac disease demonstrate a greater increase in intestinal permeability than celiacs in disease remission (5) (Figure 2). This data seems to suggest that when exposed to gliadin, everyone has some increase in intestinal permeability with those with CD and GS showing a greater response than those without CD or in remission from CD. Whether this actually leads to any health outcomes is still largely unknown.
The Culprits
One of the critical aspects of both celiac and NCGS is the antigens/problematic proteins/problematic compounds themselves. There are essentially two major things the research has highlight as culprits: gluten proteins (mainly gliadin) and FODMAPS.
Gluten Proteins
For this article (solely for exploration of an idea) let us grant a major assumption: NCGS is a result of the exposure to undigested gluten proteins to the intestinal tract that results in a pathological response. There is an alternative hypothesis that it is solely FODMAPS, but let us just stick with gluten for now. This assumption is perhaps the most widely accepted hypothesis about NCGS. I would contend that granting this assumption is wildly generous, but for this article I believe we can grant this and still sort through this topic with nuance and highlight key concepts about why I think this may be a piece to the story but perhaps not the whole story.
Gluten proteins, mainly gliadin, are often believe to be “indigestible” due to their proline rich structure. This allows them to be exert their “badness” in the intestinal tract with varying degrees of efficacy based on genetic and other factors. It appears that gluten proteins can be digested in humans; interestingly in both healthy people and those with celiac (6, 7). Now this does not completely exclude the idea that short regions of the protein may not be fully digested and still exert detrimental effects on the human gut. However, it does indicate that gluten proteins are indeed digested. It may be that the amount to which these proteins are digested and how much makes it to the intestine determines what type of response, if any, people have to gluten. However, as we will see below it may be the actual digestion of gluten that causes a problem.
The Microbiome: The Human-Environment Interface.
Gluten and the Microbiome
“Approximately 95% of the patients inherit the alleles encoding for the HLA-DQ2 and HLA-DQ8 molecules, but only a small percentage develops CD [8]. Studies of identical twins have also shown that one twin did not develop CD in 25% of the cases studied [9], supporting the role played by environmental factors in the aetiology of this disorder.”
In addition to personal variation of the amount of pepsin and trypsin produced and the digestion of gluten by these enzymes, the microbiome may play a substantial role in the digestion of gluten. Despite the studies cited above, a mixture of undigested proteins and peptides is available for bacterial metabolism in the gastrointestinal tract. As the microbiome is an integral part of the interface between the inside of our bodies and the environment, aka the human gut, it is likely that the microbiome plays an important role in CD and NCGS. Indeed, There are several studies showing that the human intestine exhibits a large variety of bacteria capable of utilizing gluten proteins and peptides as nutrients, essentially breaking down the proteins to use for their own metabolism (10, 11, 12).
Now it is tempting for one to make the following conclusion: individuals with higher levels of bacteria that fully digest gluten, thus having less gluten based proteins present, will have lower rates of CD and NCGS. This is the natural hypothesis that arises from the data above. Fortunately, science is never straight forward and always more interesting than we initially imagine. In a very interesting experiment a group of scientists examined people with and without CD and showed that bacteria and their enzymes that breakdown gluten were only present in individuals with CD, not in those without CD (13). Thus induction of gliadin proteolysis (gliadin break down) in the human gut might not be the solution, but the origin of CD. One of the proposed mechanisms in this study was that the type of bacteria that feed on gluten attack mucosal linings and degrade the intestinal lumen. As it currently stands both hypothesis seems to present some compelling arguments and we can look at the data from two perspectives:
- Digestion of gluten by bacteria contributes to celiac disease and NCGS via these bacteria metabolizing gluten and attacking the intestinal mucosa and disrupting the integrity of the gut barrier.
- Digestion of gluten by bacteria reduces the amount of reactive peptides in the intestinal track and help to ameliorate gliadin induced increases in gut permeability.
Now on the surface these seem mutually exclusive, but they may not be as the type and amount of bacteria in your gut may determine which of these scenarios is occurring. If you are content with this surface level explanation than you can skip to the last section, “How to Think About Gluten”. However, if you want a better, more thorough answer. A much deeper dive on this topic is needed. While complex, this will definitely square away a lot of the research. So down the rabbit hole we go.
Part 3: Even Harder Science
Down the Digestive Rabbit Hole: A Deep Dive on the Microbiome and Gluten
Mammalian digestive enzymes are only partly capable of cleaving gluten. Fragments of undigested gluten induce toxic responses in celiac patients (14). The major human gastrointestinal proteases, such as pepsin, trypsin, chymotrypsin, carboxypeptidases A and B, elastases and brush-border membrane enzymes of the small intestine are unable to cleave certain immunogenic gluten peptides, due to a lack of post-proline cleavage-site specificity (14). For example, the 33-mer peptide derived from α-gliadins is resistant to human intestinal proteases, and also stimulate CD4+ T-cells in the lamina propria of HLA-DQ2-positive celiac disease (CD) patients (15).
For this reason it is often assumed, and sometimes defended fanatically by certain “groups”, that humans cannot digest gluten, and therefore no single human (with or without CD) should be eating gluten-containing food, such as wheat. For them the story ends here. But there is a lot more to it.
The gastrointestinal microbiome and intestinal dysbiosis in CD
The rapidity of the increase in disease rates could never be solely explained by changes in genetic make-up (16). For example, CD autoimmunity doubled between 1974 thus excluding the genetic component as the cause of this increased prevalence (17). Alterations in gut microbiota can trigger increased immune stimulation, epithelial dysfunction and enhanced mucosal permeability (18,19) and predispose to the development of autoimmune disorders, specially food-related disease (20).
Non-human gluten-degrading proteases are naturally present in the upper human gastro-intestinal tract. These proteases are produced by the wealth of bacteria present in the oral cavity and duodenum (14,21,22). A large variety of bacteria capable of digesting gluten and use it as nutrients has been observed in the human intestine (23). Gluten can also be digested by pepsin and trypsin under physiological condition (24).
Intestinal dysbiosis has been detected in celiac disease patients. This intestinal dysbiosis is characterized by increased Gram-negative bacteria and reduced bifidobacteria (25). Factors such as milk-feeding type, duration of breast-feeding, and gastrointestinal infections, are known to influence the composition of the gut microbiota with possible relevance to celiac disease (26,27,28).
While ingested gluten is a necessary trigger for CD development, gluten reactivity alone cannot explain the absence of disease in most subjects with HLA-DQ2 and −DQ8 phenotypes, suggesting a role for the gastrointestinal microbiome (25). The overgrowth of potentially pathogenic bacteria and infections have been suggested to contribute to CD pathogenesis. In a study in free-germ rats it was apparent that the microbiome composition (potential (E. coli CBL2) and pathogenic (Shigella CBD8) enterobacteria isolated from CD patients) could play a role in the switch from tolerance to an inflammatory immune response to gluten, by altering the permeability of the intestinal mucosa (29). These enterobacteria aggravated the adverse effects of CD triggers and contributed to reducing even more goblet cell numbers and also induced massive mucus secretion.
Another study observed a distinctive ‘microbial signature’ in composition in subjects genetically susceptible to CD (30).The amount and quality of ingested gluten, together with the pattern of infant feeding and the age at which gluten is introduced in the diet might influence the risk of CD occurrence. Infants genetically susceptible to CD who are exposed to gluten early mount an immune response against gluten and develop CD autoimmunity more frequently than at-risk infants in which gluten exposure is delayed until 12 months of age (30). A lack of maturation of the gut microbiota was observed within the first 2 years of life in infants at risk of CD characterized by a relative absence of Bacteroidetes and a parallel high abundance of Firmicutes, a characteristic that remains at 24 months of age (31).
The early introduction of gluten and the lack of maturity in the GI microbiota could trigger or accelerate the development of autoimmunity. Therefore the gut microbiome between these children and children from healthy mothers (32) differ, and the composition is also different than healthy adults (31,33-38). In particular one study observed a significantly higher number of Gram-negative and potentially pro-inflammatory bacteria associated with CD (36), and another study observed increased levels of total SCFA and acetic, valeric, and butyric acids (38). Indeed it was shown that bacterial strains belonging to Bifidobacterium and Bacteroides fragilis are capable of digesting gliadin-derived peptides (39,40).
As mentioned before, emerging evidence indicate that the oral cavity is colonised with microorganisms that produce proteases capable of hydrolysing peptides rich in proline and glutamine residues (14,33,41). Another study described a faecal glutenasic activity related to diet-ingested gluten most likely derived from bacterial metabolism (42). Of 144 bacterial strains that could be involved in the metabolism of gluten proteins in the human gut, 73% belonged to Firmicutes, 15% to Actinobacteria and 12% to Proteobacteria (gram-negative bacteria) (23). Firmicutes were mainly from the genera Lactobacillus, Streptococcus, Staphylococcus and Clostridium. Several Lactobacillus strains have the ability to completely hydrolyse the 33-mer peptide, and it is known that a gluten-free diet consumed by healthy volunteers and patients with coeliac significantly affects lactobacillus populations (43,44).
Four different bacterial strains belonging to Bifidobacterium are able to metabolize gluten (23), and a gluten-free diet also appears to reduce the diversity and amount of Bifidobacterium species (43,44). A specific gliadinase pattern in duodenal samples from patients with CD was once described (45), and Bifidobacterium species, such as B. longum, B. animalis and B. bifidum, of human origin can digest gliadin peptides and inhibit the inflammatory response induced by gliadins (49). B. longum IATA-ES1 is able to hydrolyse the immunogenic peptide 33-mer and modulate an immune response (46).
Generally Lactic acid bacteria (LAB) account for 39% of the strains isolated from human faeces and these bacteria are also able to metabolize gluten (23). Proteases from LAB play an important role in the digestion of not-fully hydrolysed proteins in the human gut and may shorten long- and medium-sized peptides, particularly those peptides derived from dairy proteins (23,47). A higher LAB diversity was found in patients with treated coeliac disease and controls than in patients with active coeliac disease but species that showed significant differences between groups were food-related bacteria (48). Bacteroides diversity was higher in controls than in patients with active and treated coeliac disease, but Bacteroides dorei was more common in patients with active coeliac disease than in those with treated coeliac disease and control children. Conversely Bifidobacterium diversity was higher in patients with coeliac disease than in controls, specificaly Bifidobacterium adolescentis and Bifidobacterium animalis subsp lactis (48). Weissella spp and Lactobacillus fermentum were more frequently in patients with treated coeliac disease than in controls and patients with active coeliac disease.
One important point is that not all the bacteria involved in gluten metabolism are health promoting. Bacterial proteases from Staphylococcus epidermidis, Enterococcus faecalis, Escherichia coli, Clostridium perfringens and C. sordellii, may be related to inflammatory bowel disease (23,49,50).
The bacterial groups related to gluten metabolism that are altered in patients with CD include Bifidobacterium, Lactobacillus, Bacteroides, Staphylococcus, Clostridium and Escherichia coli (25,34,51). Bacteroides (34,52) and Clostridium leptum groups are more abundant in faeces and biopsies of CD patients than in controls regardless of the stage of the disease. E coli and Staphylococcus counts are also higher, but their levels were normalised after treatment with a GFD (33). Bifidobacterium levels are lower in faeces of both groups of CD patients and in biopsies of untreated CD patients, suggesting that either Bifidobacterium could protect against CD, or inherent features of the CD intestine influence Bifidobacterium colonization (3). In another study Clostridium histolyticum, C. lituseburense and Faecalibacterium prausnitzii group proportions were less abundant (51)
Mucosal immune response through IgA secretion constitutes a first line of defence responsible for neutralizing noxious antigens and pathogens. IgA, IgG and IgM-coated faecal bacterial levels were significantly lower in both untreated and treated CD patients than in healthy controls. Gram-positive to Gram-negative bacteria ratio was significantly reduced in both CD patients compared to controls (52). In these patients, reduced IgA-coated bacteria is associated with intestinal dysbiosis suggesting the existence of a barrier defect, which fails to stabilize the gut microbiota and prevent the host from the invasion of harmful antigens and pathogens (51).
| Phylum | Species | Gluten hydrolysis |
| Firmicutes | Enterococcus faecalis | ++ |
| Lactobacillus mucosae | MCG1,2,3 | + |
| Lactobacillus gasseri | MCG3 | +/− |
| Pediococcus acidilactici | MCG3 | ++ |
| Bacillus licheniformis | MCG3 | ++ |
| Bacillus subtilis | MCG1 | ++ |
| Bacillus pumilus | MCG1 | ++ |
| Paenibacillus jamilae | MCG1 | ++ |
| Clostridium botulinum/sporogenes | MCG2,3 | ++ |
| Clostridium perfringens | MCG2,3 | ++ |
| Clostridium sordellii | MCG2 | ++ |
| Clostridium butyricum/beijerinckii | MCG2 | ++ |
| Propionibacterium acnes | MCG3 | ++ |
| Stenotrophomonas maltophilia | MCG1 | + |
Table 2. Isolated bacteria from human faeces (23).
Gliadin proteolysis
Some authors propose that the induction of gliadin proteolysis in the human gut might not be the solution but the origin of CD, since gliadinases are CD specific and not of leucocyte origin (46). Gliadin-degrading protease (gliadinase) pattern is found in almost all samples from patients with CD regardless of GFD treatment and remains nearly absent in non-CD samples. Gliadinases might have a bacterial origin within the duodenum of patients with CD.
As noted before, Bifidobacterium species can digest gliadin peptides (39). These gliadin-metabolising bacteria may represent one of the environmental missing links in the development of CD, and could be absent, or present to a much lower degree, in the duodenum of all non-predisposed individuals, as compared with those patients who develop CD (45).This possible role of duodenal bacteria in the pathogenesis of CD has been described (36,52,53).
A rod-shaped bacteria were frequently associated with the mucosa of CD patients, with both active and inactive disease, but not with controls (54). Clostridium spp., Prevotella spp., and Actinomyces spp. were identified as the main components, and an important CD risk factor (54). E. coli and Staphylococcus seemed to be associated with the active phase of the disease and their increases could be a secondary consequence of the inflammatory milieu trigger by gluten ingestion (34). Increased levels of Staphylococcus in duodenal biopsies were also detected (34,55). In another study, increased levels of Staphylococcus and Enterobacteriaceae in infants with allergies suggested a relationship between these bacterial groups and immune dysregulation (56).
In CD the mucosal tolerance to the gut microbiota is deregulated possibly due to a higher percentage of IgA-coated Bifidobacterium than IgA-coated Bacteroides-Prevotella. This results in an increased interaction between the gut mucosal immune system and this bacterial group, which contributes to mucosal tolerance towards high gut Bifidobacterium concentrations (52,57).
The reductions in beneficial Gram-positive bacteria could favor the residence and interactions of harmful Gram-negative bacteria within the mucosal surface thereby contributing to loss of gluten tolerance (53). An increased proportion of Bacteroides-Prevotella group coupled with a weaker defensive IgA response could explain the recurrent relationship found between Bacteroides and inflamed gut mucosa in CD (53,34,36). A study observed increased levels of total SCFA and acetic, valeric, and butyric acids in CD patients (38). Increased butyric acid production could be a common feature of patients and relatives (60), since first degree CD relatives only have higher levels butyric acid but not higher levels of acetic acids and total SCFAs (39).
Small-intestinal bacterial overgrowth or SIBO is also known to affect most CD patients, with persistence of gastrointestinal symptoms after gluten withdrawal (25) and is found in both symptomatic treated or untreated CD (60). While increased numbers of enterobacteria or staphylococci may be are secondary consequences of the disease, increased Bacteroides species numbers and virulent-E. coli clones and decreased Bifidobacterium species numbers are associated with CD, regardless of symptoms and inflammation and, therefore, could play a more prominent role in this disorder (25).
The glycocalyx/mucous layer of the jejunal mucosa has unique carbohydrate structures that could modify the specificity of bacterial adhesion. These structures were found to be are altered in CD patients (25,53,61), suggesting that either a particular glycosylation pattern in predisposed individuals favors harmful bacterial adhesion, which contributes to CD pathogenesis (25,53) or modifications in the composition of the intestinal microbiota lead to alterations in the glycosylation pattern and its defensive role of the mucus layer against infections and CD (25).
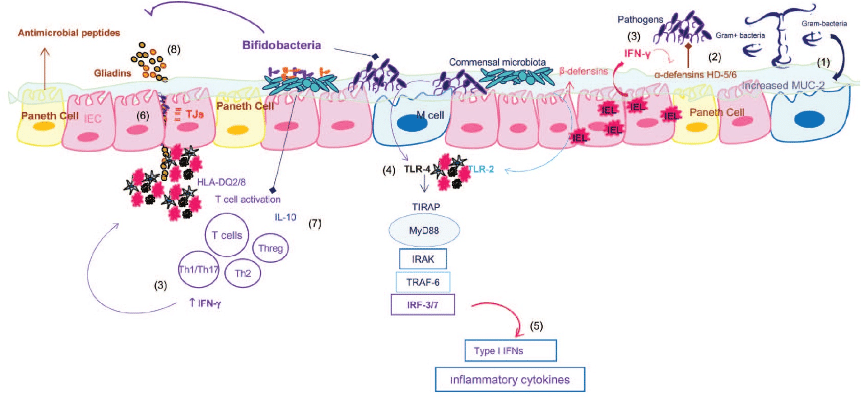
Schematic representation of the pathogenic mechanism underlying celiac disease, and key points of possible interaction with the microbiota and bifidobacteria (12).
- (1) Expression of mucin-2 (MUC2) is significantly increased, probably in response to high IFN-γ production by intraepithelial lymphocytes and secondarily to the overgrowth of potentially pathogenic bacteria in active CD patients; the glycosylation pattern of themucus layer is also modified in active and nonactive CD, and these aspects can also be modulated by the microbiota and predispose to CD.
- (2) Expression of α-defensins HD-5 and HD-6 by Paneth cells is increased in active CD patients, probably in response to high IFN-γ production by intraepithelial lymphocytes and secondarily to the overgrowth of potentially pathogenic bacteria in active CD patients.
- (4) Expression of TLR4 is increased in CD patients and TLR signaling involved in the response to commensals and pathogens.
- (5) TLR4 signaling can lead to activate interferon regulatory factor 3 (IRF3) or IRF7 leading to the production of type I IFNs that stimulate IFN-γ, already overproduced in the aberrant response to gluten.
- (6) CD patients present increased intestinal permeability due to alterations in distribution and expression of tight junction (TJ) and TJ-associated proteins.
- Specific bifidobacterial strains could play a protective role in CD pathogenesis, by increasing TJ expression of intestinal epithelial cells and reducing paracellular permeability and thus preventing/limiting gliadin translocation to the lamina propria and the consequent inflammatory response; by regulating the inflammatory effects of the altered microbiota via production of anti-inflammatory cytokines (IL-10) and reduction of IFN-γ ; by contributing to hydrolyzing gliadin peptides and thus reducing their toxicity on epithelial cells; and by increasing the number of goblet cells producing mucus and enhancing the production of inhibitors of metalloproteases (TIMP-1), which protect against tissue damage.
Defensins
Defense are proteins that contribute to host defense. Expression of α-defensins HD-5 and HD-6 in Paneth cells exceptionally high in the human small intestine (62). These defensins have antimicrobial activity and contribute to create a hostile environment that prevents overgrowth of bacteria and infections (63).
In CD patients, increased expression of α-defensins HD-5 and HD-6 would appear to be a secondary consequence of the inflammatory intestinal milieu characteristic of the disease linked to an overgrowth of some potentially pathogenic bacteria (E. coli), since expression returns to normal levels in patients under a gluten-free diet (25). β-Defensins are inducible antimicrobial peptides mainly produced by epithelial cells and they also play a crucial role in the innate immune system (64).
High copy numbers (more than 4) of β-defensins were underrepresented among CD patients, suggesting that increased copy numbers of β-defensins in CD could afford protection from CD, possibly by preventing bacterial infiltration and preserving gut epithelial integrity (25,64,65).
Therapy
Aside from a gluten-free diet, gluten-degrading enzymes are being pursued as adjunctive therapeutics for celiac disease, proline and glutamine-specific endopeptidases from bacteria, fungi and barley are currently being explored (14,66-72). The gluten-degrading enzymes from the oral bacteria (mostly a harmless residents) could be isolated and further pursued as a pharmaceutical drug, or dietary supplement (14). The gluten-degrading bacteria themselves could be developed as probiotic agents to achieve gluten digestion. B. longum IATA-ES1 is commercially available in food as a probiotic bacterium for patients with CD (14).
The Microbiome Story Summarized.
It appears there is a large variety of bacteria capable of digesting gluten with gluten-degrading proteases naturally present in the upper human gastro-intestinal tract, indicating humans can and do digest gluten. The oral cavity is colonised with microorganisms that produce proteases capable of hydrolysing peptides rich in proline and glutamine residues such as those found in gluten proteins. It is quite clear that bacterial groups related to gluten metabolism are altered in patients with CD. Intestinal dysbiosis is present in celiac disease patients, characterized by increased Gram-negative bacteria, other potentially pro-inflammatory bacteria and reduced bifidobacteria. Small-intestinal bacterial overgrowth (SIBO) and infections have been suggested to contribute to CD pathogenesis with persistence of gastrointestinal symptoms after gluten withdrawal. Pathogenic enterobacteria could play a role in the switch from tolerance to an inflammatory immune response to gluten, by altering the permeability of the intestinal mucosa.
It appears that a lack of maturation of the gut microbiota is observed within the first 2 years of life in infants at risk of CD and that the early introduction of gluten and the lack of maturity in the GI microbiota could trigger or accelerate the development of autoimmunity. Reduced IgA-coated bacteria is associated with intestinal dysbiosis suggesting the existence of a barrier defect, which fails to stabilize the gut microbiota and prevent the host from the invasion of harmful antigens and pathogens. Lastly, either Bifidobacterium could protect against CD, or inherent features of the CD intestine influence Bifidobacterium colonization.
Part 4: How To Think About Gluten
Out of the rabbit hole and up into the clouds.
If you poll people on how they think about gluten they usually fall somewhere on the spectrum of “Gluten is Satan” to “Eating Gluten Free is for Idiots” (Ironically, a vegetarian protein option made entirely from gluten protein is called seitan and pronounced say-tan, just like satan).
In order to think logically about gluten we need to separate our ideas from who we are, and remove the emotional aspect of this discussion. Remember, it is ok to be wrong and change your stance on an issue. Personally, I waffle over where I fall on this issue probably weekly. But let’s review what we have covered so far and present some additional data and ways to think about the issue.
Now we need to discuss some additional data. First we can dissect down into who benefits from a gluten free diet. If we stratify people based on celiac diagnosis vs. no celiac diagnosis and based on their response or lack of response to gluten we can then label these groups based on whether the evidence supports a therapeutic benefit of eating gluten free (Table 3)
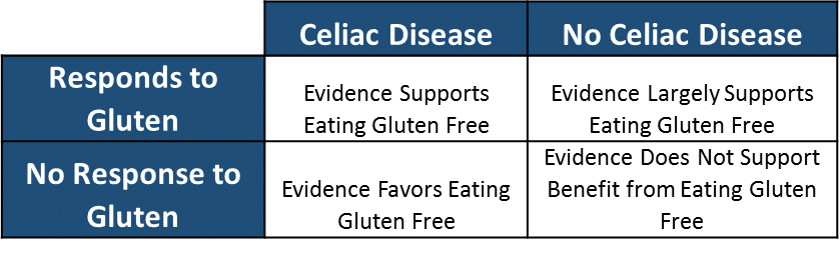
Now we can overlay these groups with the figures from table one and calculate: 1) the number of people that evidence supports eating gluten free, and 2) the number of people that evidence does not eating gluten free.
Number of People that Evidence Supports the Benefit of Eating Gluten Free: 24,478,050
Number of People that Evidence Does Not Support the Benefit of Eating Gluten Free: 293,521,950
This suggests that approximately 25 million people likely have some legitimate reason to avoid gluten based on the cumulative evidence to date.
Let us then ask the next logical question. Who is likely to be harmed from removing gluten from their diet based on the same categorical stratification and calculate: 1) the number of people that are likely to be harmed eating gluten free.
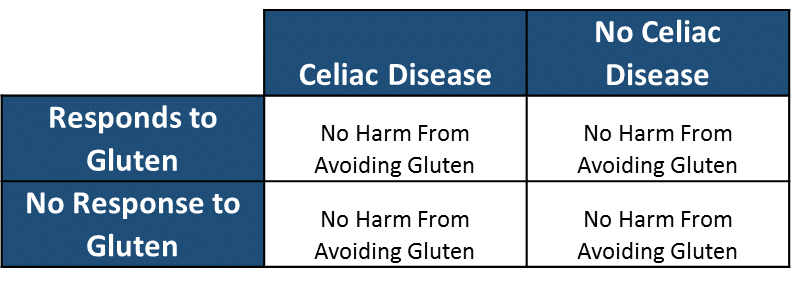
Number of People that are Likely to be Harmed Eating Gluten Free: 0
Here is currently how I view the “gluten issue”. Celiac disease presents and interesting autoimmune condition in which gluten exposure presents a substantial health detriment both acutely and chronically. Individuals without CD but who consistently and repeatedly report adverse reactions to gluten (anywhere along the spectrum) are likely to benefit from restricting gluten from their diet. While there are some interesting hypothesis about the effect of gluten intake and chronic diseases (e.g. Alzheimer’s), the data is not substantial enough to warrant guidelines recommending the restriction of gluten to people who are not celiac or who do not present with adverse reactions to gluten exposure.
Is this rise of glutensanity the worst thing? Probably not. There are likely a lot of people who would not have known to attribute their GI issues to gluten as it was not a conversation being had a dinner tables, bars (drinking gluten free beer of course), or gym locker rooms until more recently. The abuse of marketing to drive profit is an issue, but then again what do you expect, marketers ruin everything.
*I would conjecture this number is artificially high. The data showing these ranges of percentages are from research studies in which many of the patients are self-selected. It is very likely that the actual rate of NCGS is lower, perhaps 2-4% of the entire population. Regardless, it does not detract from the point made in any appreciate sense.
References
- Gujral N, Freeman HJ, Thomson AB. Celiac disease: prevalence, diagnosis, pathogenesis and treatment. World journal of gastroenterology. 2012;18:6036-6059.
- https://celiac.org/celiac-disease/resources/checklist/
- Drago S, El Asmar R, Di Pierro M, Grazia Clemente M, Tripathi A, Sapone A, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scandinavian journal of gastroenterology. 2006;41:408-419.
- Lammers KM, Lu R, Brownley J, Lu B, Gerard C, Thomas K, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008;135:194-204 e193.
- Hollon J, Puppa EL, Greenwald B, Goldberg E, Guerrerio A, Fasano A. Effect of gliadin on permeability of intestinal biopsy explants from celiac disease patients and patients with non-celiac gluten sensitivity. Nutrients. 2015;7:1565-1576.
- Bruce G, Woodley JF, Swan CH. Breakdown of gliadin peptides by intestinal brush borders from coeliac patients. Gut. 1984;25:919-924.
- Jonsson T, Memon AA, Sundquist K, Sundquist J, Olsson S, Nalla A, et al. Digested wheat gluten inhibits binding between leptin and its receptor. BMC biochemistry. 2015;16:3.
- Mearin ML, Ivarsson A, Dickey W. Coeliac disease: is it time for mass screening? Best practice & research. Clinical gastroenterology. 2005;19:441-452.
- Greco L, Romino R, Coto I, Di Cosmo N, Percopo S, Maglio M, et al. The first large population based twin study of coeliac disease. 2002;50:624-628.
- Caminero A, Herran AR, Nistal E, Perez-Andres J, Vaquero L, Vivas S, et al. Diversity of the cultivable human gut microbiome involved in gluten metabolism: isolation of microorganisms with potential interest for coeliac disease. FEMS microbiology ecology. 2014;88:309-319.
- Zamakhchari M, Wei G, Dewhirst F, Lee J, Schuppan D, Oppenheim FG, et al. Identification of Rothia bacteria as gluten-degrading natural colonizers of the upper gastro-intestinal tract. PloS one. 2011;6:e24455.
- Fernandez-Feo M, Wei G, Blumenkranz G, Dewhirst FE, Schuppan D, Oppenheim FG, et al. The cultivable human oral gluten-degrading microbiome and its potential implications in coeliac disease and gluten sensitivity. Clinical microbiology and infection : the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2013;19:E386-394.
- Bernardo D, Garrote JA, Nadal I, León AJ, Calvo C, Fernández-Salazar L, et al. Is it true that coeliacs do not digest gliadin? Degradation pattern of gliadin in coeliac disease small intestinal mucosa. Gut. 2009;58:886-887.
- Martin Fernandez-Feo, Guoxian Wei, Gabriel Blumenkranz, Floyd E. Dewhirst, Detlef Schuppan, Frank G. Oppenheim and Eva J. Helmerhorst. The Cultivable Human Oral Gluten-Degrading Microbiome and its Potential Implications in Celiac Disease and Gluten Sensitivity. Clin Microbiol Infect. 2013 Sep; 19(9): E386–E394.
- Vader LW, de Ru A, van der Wal Y, et al. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. The Journal of experimental medicine. 2002;195:643–649. [PMC free article] [PubMed]
- Noverr MC, Huffnagle GB. Does the microbiota regulate immune responses outside the gut? Trends Microbiol. 2004;12:562–568. [PubMed]
- Catassi C, Kryszak D, Bhatti B, Sturgeon C, Helzlsouer K, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Annals of medicine. 2010;42:530–538. [PubMed]
- Penders J, Stobberingh EE, van den Brandt PA, Thijs C. The role of the intestinal microbiota in the development of atopic disorders. 2007;62:1223–1236. [PubMed]
- Sartor RB. Microbial influences in inflammatory bowel diseases. 2008;134:577–594. [PubMed]
- Kranich J, Maslowski KM, Mackay CR. Commensal flora and the regulation of inflammatory and autoimmune responses. Seminars in immunology. 2011;23:139–145. [PubMed]
- Zamakhchari M, Wei G, Dewhirst F, et al. Identification of rothia bacteria as gluten-degrading natural colonizers of the upper gastro-intestinal tract. PloS one. 2011;6:e24455. [PMC free article] [PubMed]
- Helmerhorst EJ, Zamakhchari M, Schuppan D, Oppenheim FG. Discovery of a novel and rich source of gluten-degrading microbial enzymes in the oral cavity. PloS one. 2010;5:e13264. [PMC free article] [PubMed]
- Caminero A, Herrán AR, Nistal E, Pérez-Andrés J, Vaquero L, Vivas S, Ruiz de Morales JM, Albillos SM, Casqueiro J. Diversity of the cultivable human gut microbiome involved in gluten metabolism: isolation of microorganisms with potential interest for coeliac disease. FEMS Microbiol Ecol. 2014 May;88(2):309-19. doi: 10.1111/1574-6941.12295. Epub 2014 Mar 3.
- Tommy Jönsson, Ashfaque A Memon, Kristina Sundquist, Jan Sundquist, Stefan Olsson, Amarnadh Nalla, Mikael Bauer, and Sara Linse. Digested wheat gluten inhibits binding between leptin and its receptor. BMC Biochem. 2015; 16: 3.
- Yolanda Sanz, Giada De Palma, and Moisés Laparra. Unraveling the Ties between Celiac Disease and Intestinal Microbiota. International Reviews of Immunology, 30:207–218, 2011
- Ivarsson A, Hernell O, Stenlund H, Persson LA. Breast-feeding protects against coeliac disease. Am J Clin Nutr. 2002;75: 914–921.
- Stene LC, Honeyman MC, Hoffenberg EJ, et al. Rotavirus infection frequency and risk of coeliac disease autoimmunity in early childhood: a longitudinal study. Am J Gastroenterol., 2006;101:2333–2340.
- Akobeng AK, Ramanan AV, Buchan I, Heller RF. Effect of breast feeding on risk of coeliac disease: a systematic review and meta-analysis of observational studies. Arch Dis Child. 2006;91:39–43.
- Cinova J, De Palma G, Stepankova R, et al. Role of intestinal bacteria in gliadin-induced changes in intestinal mucosa: Study in germ-free rats. PloS one. 2011;6:e16169. [PMC free article] [PubMed]
- Schippa S, Iebba V, Barbato M, Di Nardo G, Totino V, et al. A distinctive ‘microbial signature’ in celiac pediatric patients. BMC microbiology. 2010;10:175. [PMC free article] [PubMed]
- Sellitto M, Bai G, Serena G, et al. Proof of concept of microbiome-metabolome analysis and delayed gluten exposure on celiac disease autoimmunity in genetically at-risk infants. PloS one. 2012;7:e33387. [PMC free article] [PubMed]
- Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007;5:e177. [PMC free article] [PubMed]
- Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Imbalances in faecal and duodenal Bifidobacterium species composition in active and non-active coeliac disease. BMC Microbiol. 2008;8:232. [PMC free article] [PubMed]
- Collado MC, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Specific duodenal and faecal bacterial groups associated with paediatric coeliac disease. J Clin Pathol. 2009;62:264–269. [PubMed]
- Di Cagno R, Rizzello CG, Gagliardi F, Ricciuti P, Ndagijimana M, et al. Different fecal microbiotas and volatile organic compounds in treated and untreated children with celiac disease. Appl Environ Microbiol. 2009;75:3963–3971. [PMC free article] [PubMed]
- Nadal I, Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Imbalance in the composition of the duodenal microbiota of children with coeliac disease. J Med Microbiol. 2007;56:1669–1674. [PubMed]
- Sanz Y, Sanchez E, Marzotto M, Calabuig M, Torriani S, et al. Differences in faecal bacterial communities in coeliac and healthy children as detected by PCR and denaturing gradient gel electrophoresis. FEMS Immunol Med Microbiol. 2007;51:562–568. [PubMed]
- Tjellstrom B, Stenhammar L, Hogberg L, Falth-Magnusson K, Magnusson KE, et al. Gut microflora associated characteristics in children with celiac disease. Am J Gastroenterol. 2005;100:2784–2788. [PubMed]
- Laparra JM & Sanz Y (2010) Bifidobacteria inhibit the inflammatory response induced by gliadins in intestinal epithelial cells via modifications of toxic peptide generation during digestion. J Cell Biochem 109: 801–
- Sanchez E, Laparra JM & Sanz Y (2012) Discerning the role of Bacteroides fragilis in celiac disease pathogenesis. Appl Environ Microbiol 78: 6507–
- Helmerhorst EJ, Zamakhchari M, Schuppan D & OppenheimFG (2010) Discovery of a novel and rich source of gluten-degrading microbial enzymes in the oral cavity. PloS One 5: e13264.
- Caminero A, Nistal E, Arias L et al. (2012). A gluten metabolism study in healthy individuals shows the presence of faecal glutenasic activity. Eur J Nutr 51: 293–
- De Palma G, Nadal I, Collado MC, Sanz Y (2009). Effects of a gluten-free diet on gut microbiota and immune function in healthy adult human subjects. Br J Nutr 102: 1154–1160.
- Nistal E, Caminero A, Vivas S, Ruiz de Morales JM, Sáenz de Miera LE, Rodríguez-Aparicio LB, Casqueiro J (2012). Differences in faecal bacteria populations and faecal bacteria metabolism in healthy adults and celiac disease patients. Biochimie 94: 1724–1729
- Bernardo D, Garrote JA, Nadal I, Le_on AJ, Calvo AJ, Fernandez-Salazar L, Blanco-Quiros A, Sanz Y & Arranz E (2009). Is it true that coeliacs do not digest gliadin? Degradation pattern of gliadin in coeliac disease small intestinal mucosa. Gut 58: 886–887
- Laparra JM, Olivares M, Gallina O, Sanz Y (2012). Bifidobacterium longum CECT 7347 modulates immune responses in a gliadin-induced enteropathy animal model. PLoS One 7: e30744.
- Pessione E (2012) Lactic acid bacteria contribution to gut microbiota complexity: lights and shadows. Front Cell Infect Microbiol 2: 86.
- Sanchez E,Donat E, Ribes-Koninckx C, Calabuig M, Sanz Y. Intestinal Bacteroides species associated with coeliac disease. J Clin Pathol. 2010;63: 1105–1111.
- PruteanuM, HylandNP, ClarkeDJ, KielyB, ShanahanF (2011). Degradation of the extracellular matrix components by bacterial-derived metalloproteases: implications for inflammatory bowel diseases. Inflamm Bowel Dis 17: 1189–1200. CrossRef Medline
- Steck N, Mueller K, Schemann M & Haller D (2012). Bacterial proteases in IBD and IBS. Gut 61: 1610–
- De Palma G, Nadal I, Medina M, Donat E, Ribes-Koninckx C, Calabuig M & Sanz Y (2010). Intestinal dysbiosis and reduced immunoglobulin-coated bacteria associated with coeliac disease in children. BMC Microbiol 10: 63. CrossRefMedline
- Tursi A, Brandimarte G, Giorgetti G. High prevalence of small intestinal bacterial overgrowth in celiac patients with persistence of gastrointestinal symptoms after gluten withdrawal. Am J Gastroenterol 2003;98:839–43.
- Forsberg G, Fahlgren A, Horstedt P, et al. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am J Gastroenterol 2004;99:894–904.
- Ou G, Hedberg M, H¨orstedt P, et al. Proximal small intestinal microbiota and identification of rod shaped bacteria associated with childhood CD. AmJ Gastroenterol. 2009;104: 3058–3067.
- Collado MC, Calabuig M, Sanz Y. Differences between the faecal microbiota of coeliac children and healthy controls. Curr Issues Intest Microbiol 2007;8:9–14.
- Bjorksten B, Sepp E, Julge K, et al. Allergy development and the intestinal microflora during the first year of life. J Allergy Clin Immunol 2001;108:516–20.
- van der Waaij LA, Kroese FG, Visser A, Nelis GF, Westerveld BD, Jansen PL, Hunter JO: Immunoglobulin coating of faecal bacteria in inflammatory bowel disease. Eur J Gastroenterol Hepatol 2004, 16:669-674.
- TjellstromB, Stenhammar L,H¨ogberg L, et al. Screening-detected and symptomatic untreated celiac children show similar gut microflora-associated characteristics. Scand J Gastroenterol. 2010;45:1059–1062.
- Tjellstr¨om B, Stenhammar H¨ogberg L. et al. Gut microflora associated characteristics in first-degree relatives of children with CD. Scand J Gastroenterol. 42: 2007;1204–1208.
- Rubio-Tapia A, Barton SH, Rosenblatt JE, Murray JA. Prevalence of small intestine bacterial overgrowth diagnosed by quantitative culture of intestinal aspirate in celiac disease. J Clin Gastroenterol. 2009;43: 157–161
- Vecchi M, Torgano G, Tronconi S, Agape D, Ronchi G. Evidence of altered structural and secretory glycoconjugates in the jejunal mucosa of patients with gluten sensitive enteropathy and subtotal villous atrophy. Gut. 1989;30: 804–810.
- Wehkamp J, Chu H, Shen B, et al. Paneth cell antimicrobial peptides: topographical distribution and quantification in human gastrointestinal tissues. FEBS Lett. 2006;580: 5344–5350.
- Bevins CL, Nita H, Salzman NH, Ghosh D, Huttner KM. Human defensin-5 (HD-5) transgenic mice: paneth cell expression and protection from lethal Salmonella typhimurium Gastroenterology. 2002;A169.
- Zhao C,Wang I, Lehrer RI. Widespread expression of beta-defensin hBD-1 inhumansecretory glands and epithelial cells. FEBS Lett. 1996;396: 319–322.
- Fernandez-Jimenez N, Castellanos-Rubio A, Plaza-Izurieta L. et al. Analysis of beta-defensin and Toll-like receptor gene copy number variation in CD. Hum Immunol. 2010;71: 833-836
- Mitea C, Havenaar R, Drijfhout JW, Edens L, Dekking L, Koning F. Efficient degradation of gluten by a prolyl endoprotease in a gastrointestinal model: Implications for coeliac disease. Gut. 2008; 57:25–32. [PubMed: 17494108]
- Marti T, Molberg O, Li Q, Gray GM, Khosla C, Sollid LM. Prolyl endopeptidase-mediated destruction of t cell epitopes in whole gluten: Chemical and immunological characterisation. The Journal of pharmacology and experimental therapeutics. 2005; 312:19–26. [PubMed: 15358813]
- Shan L, Marti T, Sollid LM, Gray GM, Khosla C. Comparative biochemical analysis of three bacterial prolyl endopeptidases: Implications for coeliac sprue. The Biochemical journal. 2004; 383:311–318. [PubMed: 15245330]
- Stepniak D, Spaenij-Dekking L, Mitea C, et al. Highly efficient gluten degradation with a newly identified prolyl endoprotease: Implications for celiac disease. American journal of physiology. 2006; 291:G621–G629. [PubMed: 16690904]
- Siegel M, Bethune MT, Gass J, et al. Rational design of combination enzyme therapy for celiac sprue. Chemistry & biology. 2006; 13:649–658. [PubMed: 16793522]
- Cerf-Bensussan N, Matysiak-Budnik T, Cellier C, Heyman M. Oral proteases: A new approach to managing coeliac disease. Gut. 2007; 56:157–160. [PubMed: 16950833]
- Bethune MT, Khosla C. Oral enzyme therapy for celiac sprue. Methods Enzymol. 2012; 502:241–271. [PubMed: 22208988]